
Abstrakt
Příspěvek poukazuje na význam metod softcomputingu vbioinformatice. Práce je věnována základním principům a pojmům z oblasti bioinformatiky a softcomputingu. Hlavní část je zaměřena na použití evolučních algoritmů vbioinformatice, vkteré byly prezentovány typické úlohy (zarovnávání a porovnávání DNA, RNA a proteinových sekvencí; mapování genů na chromozómech; hledání genů a identifikace promotorů ze sekvencí DNA; interpretace genové exprese a microarray dat; regulace genových sítí; konstrukce fylogenetických stromů; predikce struktury DNA, RNA a proteinů; molekulární design a molekulární dokování). Praktická část je věnována výpočtům vprogramu MEGA následné analýze jejich výstupů.
Klíčová slova: bioinformatika, sekvence DNA, evoluční algoritmus, geny.
Abstract
The entry documents the philosophy and terms from bioinformatics and softcomputings. This studies are dedicated to basic principles and concepts of bioinformatics and softcomputing. The main part contain the search focused on the use of evolutionary algorithms in bioinformatics, which were presented typical problems (alignment and comparison of DNA, RNA and protein sequences; gene mapping on chromosomes; searching for genes and identification of promoters from DNA sequences; interpretation of gene expression and microarray data; gene regulatory networks; construction of phylogenetic trees; structure prediction of DNA, RNA and proteins; molecular design and molecular docking). The practical part includes a research of examples in MEGA program with a follow-up analysis.
Keywords: bioinformatics, DNA sequences, evolutionary algorithms, genes.
Úvod
Tato práce poukazuje na význam metod softcomputingu vbioinformatice. Na počátku bylo nutné sumarizovat velké množství informací a údajů, které souvisejí sdaným tématem. Po získání, setřídění a prostudování zdrojů a dat následovala excerpce důležitých materiálů, což bylo předmětem této práce.
Bionformatika
Bioinformatika vznikla jako odnož genomiky. Věnuje se hlavně zpracováním a analýzou biologických dát získaných vědeckým výzkumem, přičemž velký důraz se klade na identifikaci a diagnostiku nových genů aproteinů a určení funkce bílkovin, které tyto geny kódují. Moderní bioinformatika se dnes používá také pro vývoj nových léků, vkriminalistice, kde se sjejí pomocí dá usvědčit pachatel trestného činu a vmnoha dalších odvětvích.
DNA
Deoxyribonukleová kyselina (DNA) a proteiny jsou makromolekuly postaveny zbiologických makromolekul lineárních řetězců základních chemických složek. DNA se skládá ze sekvencevelkého počtu sekvencí nukleotidů. DNA hraje zásadní roli vrůzných biochemických procesech živých organismů. Obsahuje šablony pro syntézu bílkovin, které jsou nezbytné pro každý organismus. Druhá role, ve které je DNA nezbytná pro život je, že slouží jako médium pro přenos dědičné informace z generace nageneraci.
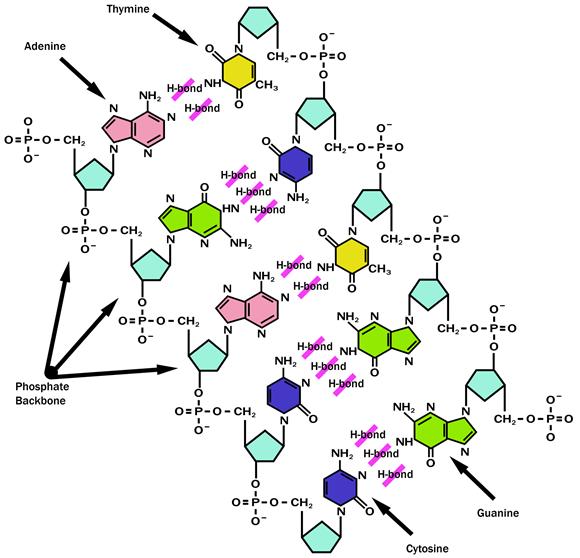
Obrázek 1 - Struktura DNA - na každé straně je vlákno tvořené fosfátovými skupinami a deoxyribózovými cukry, které se pravidelně střídají a mezi sebou jsou spojené dusíkatými bázemi.
Evoluční algoritmy
V přírodě biologičtí jedinci jedné populace mezi sebou soutěží o přežití a možnost reprodukce na základě toho, jak dobře jsou přizpůsobeni prostředí. V průběhu mnoha generací se struktura dané populace vyvíjí na základě Darwinovy teorie o přirozeném výběru a přežívání jen těch jedinců, kteří mají největší sílu (schopnost přežít neboli fitness).
Evoluční algoritmy se snaží využít modelů evolučních procesů, aby tak nalezly řešení náročných a rozsáhlých úloh. Veškeré takové modely mají několik společných rysů:
Evoluční algoritmy jsou zastřešujícím termínem pro různé přístupy využívající modely evolučních procesů pro účely nemající téměř nic společného s biologií. Snaží se využívat představy o hnacích silách evoluce živé hmoty pro účely optimalizace. Evoluční algoritmy se především používají pro řešení velkých komplexních optimalizačních problémů smnoha lokálními optimy, protože je zde menší pravděpodobnost, že uváznou v lokálním minimu než u tradičních gradientních metod. Řešení evolučními algoritmy nezávisí nagradientních informacích, a tak jsou především vhodné pro řešení problémů, kde jsou takové informace nedostupné, drahé nebo odhadnuté. Mohou řešit i problémy, které nejsou přímo vyjádřeny přesnou účelovou funkcí. Evoluční algoritmy jsou mnohem robustnější než jiné prohledávací algoritmy.
Fragment Assembly Problem
Většina problémů v bioinformatice, jako je přiřazení sekvencí, mapování genů nachromozómech, konstrukce fylogenetických stromů, stanovení struktury bílkovin, FAP (Fragment Assembly Problem - skládání fragmentů DNA) aj. se řeší podobně jako TSP (Traveling Salesman Problem - problém obchodního cestujícího), ale svýznamnými odlišnostmi, např. nehledáme okružní cestu.
Skládání fragmentů DNA je konečnou fází čtení dlouhých řetězců DNA. Dlouhé řetězce DNA jsou několikrát přeťaty a tím je vytvořena velká skupina fragmentů. Fragmenty jsou poté složeny do řetězce, který odpovídá čtené DNA. Tento proces je velmi komplikovaný, protože jak pořadí, tak i orientace se ztratí v předchozím stupni.
V tomto problému se snažíme poskládat nějaké fragmenty za sebe tak, aby celková délka řetězce byla co nejmenší. Různé délky se může dosáhnout díky tomu, že dva následující fragmenty se mohou zčásti překrývat a tak celkovou délku snížit. Jednotlivé fragmenty si můžeme představit jako části DNA, případně jako nějaké textové řetězce. Překrýváním jsou myšleny shodné znaky na konci prvního fragmentu a začátku druhého. Fragmenty, které následují po sobě, mohou mít některé znaky stejné, např. dva řetězce „CANX“ a „XLOPM“ by měly, umístěny vtomto pořadí, délku 8. Pokud bychom jejich pořadí prohodili, celková délka by vzrostla na 9, protože se nepřekrývají. Navíc se do každého fragmentu dají vkládat mezery, které se považují za shodné s jakýmkoliv znakem. Díky tomu se může dosáhnout větších překryvů a dalšího snížení celkové délky. Dále je třeba zmínit, že každý fragment je možné do řetězce vložit obráceně, tedy pozpátku. Cílem této optimalizační úlohy je najít pořadí fragmentů a jejich natočení tak, aby celková délka byla co nejmenší. Tato úloha může obsahovat lokální extrémy, do kterých původní algoritmus může snadno zapadnout a považovat je za globální optimum. Řešení implementovaná vrámci GA mohou tomuto uváznutí zabránit a pokračovat dál v hledání globálního extrému.
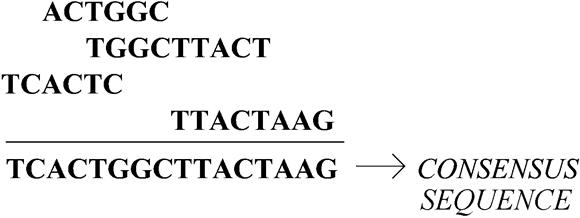
Obrázek 2 - Přiřazení fragmentů DNA
Konstrukce fylogenetických stromů pro studium evolučních vztahů
Rekonstrukce fylogeneze je vzásadě statistickým problémem, tj. procedurou kvalifikovaného odhadu. Způsoby konstrukce fylogeneze jsou v podstatě dvojí. Prvním přístupem je definování specifické sekvence kroků, neboli algoritmu, které vedou kekonstrukci stromu, druhý přístup se snaží definovat kritéria porovnání alternativních hypotéz (stromů) a rozhodnutí, která z nich je nejoptimálnější. Algoritmické metody kombinují konstrukci fylogeneze a definici té fylogeneze, která je „nejlepší“ a tudíž preferována, do jediného algoritmu. Tyto metody postupují dle stanoveného algoritmu přímočaře ke konečnému řešení, kterým je jediný fylogenetický strom. Protože u nich odpadá zdlouhavé srovnávání velkého množství vzájemně si konkurujících stromů, jsou zpravidla extrémně rychlé. Do této skupiny patří všechny metody shlukové analýzy a metoda neighbor-joining, jež sjednocuje shluky po dvojicích tak dlouho, dokud nezůstane pouze jediný shluk. Kritériem sjednocení je vzdálenost mezi dvojicí shluků a průměrná vzdálenost k ostatním uzlům v grafu.
Metody druhé skupiny, založené na stanovení kritéria optimálnosti, pracují ve dvou krocích. Vprvním kroku je definováno kritérium, podle něhož je každému stromu přiřazena určitá hodnota (skóre), která je následně použita kvzájemnému porovnání stromů. Druhý krok zahrnuje použití specifického algoritmu pro výpočet hodnoty funkce znázorňující kritérium optimálnosti a pro nalezení stromu snejlepší hodnotou této funkce. Evoluční předpoklady vprvním kroku jsou tak odděleny od matematického řešení vkroku druhém. Na rozdíl od algoritmických metod je nedílnou součástí optimalizačních metod posuzování často velkého množství alternativních stromů, které (někdy výrazně) zpomaluje dosažení konečného výsledku.
Hledání genů a podpora jejich identifikace z DNA sekvencí
Jednou z hlavních výhod tohoto přístupu je zjištění funkčnosti rodiny sekvencí smalou homologií (podobností). Genová rodina je skupina příbuzných genů, které kódují podobné bílkovinné produkty. Každá genová rodina historicky vzniká postupnou duplikací genů a jejich postupnou evolucí (na základě mutací), přičemž mohou kódované proteiny dokonce získávat nové funkce. Geny zjedné genové rodiny jsou seřazeny na jednom chromozomu, ale mohou být i rozptýleny po celém genomu naněkolika chromozomech, nebo genomické knihovně.
MEGA Software
Software MEGA (Molecular Evolutionary Genetics Analysis) se používá pro analyzování sekvenčních dat. Uplatňuje se při odhadech evoluční vzdálenosti, při tvorbě fylogenetických stromů, provádí selekční testy. V současné době již ve své páté hlavní verzi, MEGA5 obsahuje prostředky pro automatické i manuální přiřazování sekvencí, on-line dolování databází, odvození fylogenetických stromů, odhad evoluční vzdálenosti a testování evolučních hypotéz.
Příklad: Vytváření stromů ze sekvenčních dat metodou Neighbor-Joining
Průběh výpočtu:
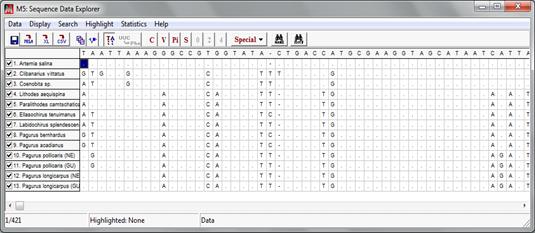
Obrázek - Datový soubor "Crab_rRNA.meg“
2) Zhlavního menu MEGA vybrat Phylogeny | Construct/Test Neighbor-Joining Tree.
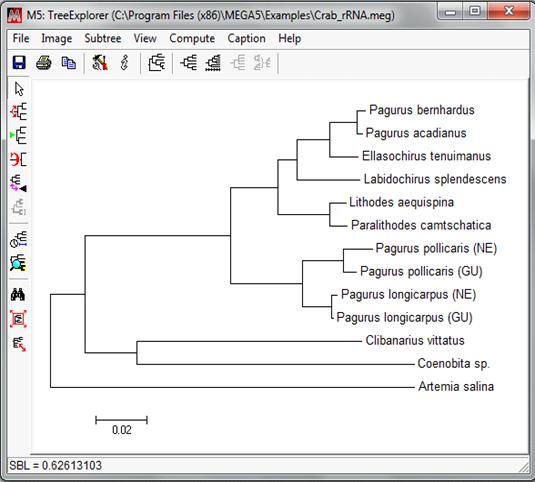
Obrázek - Zobrazení výsledného stromu
V tomto výpočtu proběhne výpočet distanční matice a pak se vykreslí fylogenetický strom, který popisuje fylogenetické vztahy mezi zadanými sekvencemi.
Závěr
Práce poukazuje na význam metod softcomputingu vbioinformatice. Smyslem práce nebylo jen shromáždit co nejvíce informací, ale představit různorodost využití metod softcomputingu zejména evolučních algoritmů v bioinformatice. Dále bych chtěl pokračovat v návrhu a vytvoření integrované sady nástrojů pro realizaci genomového projektu od assembly až po anotaci eukaryotických genomů. Tato sada nástrojů by pak měla být otestována na konkrétním genomovém projektu.
Seznam použité literatury:
Aktuální číslo
Odborný vědecký časopis Trilobit | © 2009 - 2026 Fakulta aplikované informatiky UTB ve Zlíně | ISSN 1804-1795